LCC
L’azote atmosphérique, une ressource inerte mais essentielle
L’azote moléculaire (N₂) compose près de 80 % de l’air que nous respirons. Pourtant, cette molécule est remarquablement inerte en raison de sa triple liaison très solide, ce qui la rend difficile à transformer. Dans la nature, seuls certains microorganismes sont capables de « fixer » l’azote, c’est-à-dire de le convertir en ammoniac (NH₃), une forme réactive indispensable à la fabrication des briques du vivant, comme les acides aminés. Cette transformation est assurée par des enzymes appelées nitrogénases. Depuis le XXe siècle, l’industrie a développé un procédé similaire, le procédé Haber-Bosch, qui permet de produire de l’ammoniac à grande échelle pour les engrais. Mais ce procédé est énergivore et fortement émetteur de CO₂. Inspirés par les nitrogénases, les chimistes cherchent aujourd’hui à concevoir de nouveaux matériaux et catalyseurs moléculaires capables de fixer l’azote dans des conditions plus douces et durables.
Nos recherches sur l’activation de l’azote
Activation et fonctionnalisation du diazote par des systèmes donneurs-accepteurs
Dans notre équipe, nous explorons de nouvelles stratégies hétéro-bimétalliques pour activer le diazote (N₂), inspirées du fonctionnement des nitrogénases. L’idée est d’associer un centre métallique donneur d’électrons (complexes M⁰ du groupe 6) à un acide de Lewis fort, afin de polariser la molécule et d’en révéler une réactivité inédite.
Nous avons montré que la coordination de B(C₆F₅)₃ sur un ligand N₂ induit un allongement de la liaison N≡N et une baisse significative de sa fréquence vibrationnelle. L’angle N≡N–B d’environ 140° reflète un recouvrement efficace entre l’orbitale vacante du bore et la π* du ligand diazote (Angew. Chem. Int. Ed. 2017).
Nous avons également introduit des fragments cationiques Au(I), à la fois pour leur potentiel à engager une chimie N–C originale et comme analogues isolobaux de H⁺. Des études spectroscopiques et DFT montrent que l’activation de N₂ dans ces systèmes repose sur des effets électrostatiques marqués, suggérant un rôle clé dans la réduction enzymatique de N₂ (Inorg. Chem. 2021).
Avec Al(C₆F₅)₃, bien que l’activation soit légèrement atténuée par des effets stériques, nous avons pu isoler des adduits doublement activés (M{–N≡N–Al(C₆F₅)₃}₂), inaccessibles dans la série bore. Les calculs montrent par ailleurs une réduction de l’écart HOMO–LUMO dans ces systèmes, traduisant une meilleure aptitude aux transferts d’électrons et de protons — une propriété clé pour la catalyse (Chem. Sci. 2024).
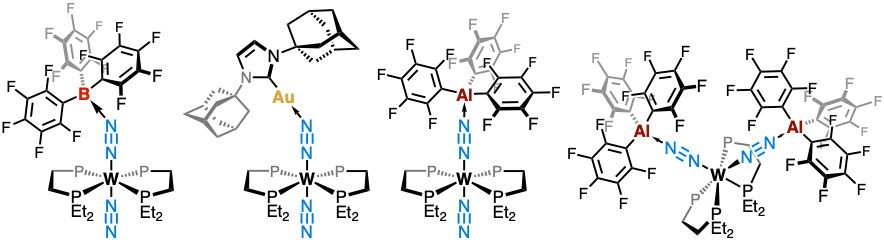
Activation de l’azote par le tungstène, combiné au bore, à l’or et à l’aluminium.
Nos travaux ouvrent ainsi la voie à une fixation homogène de N₂ assistée par acides de Lewis, où ces derniers agiraient comme co-catalyseurs dans des cycles redox doux, plus proches des conditions naturelles.
Nos adduits M–N₂/acide de Lewis se comportent comme des paires de Lewis frustrées (PLF), capables d’activer des liaisons fortes en conditions douces. En traitant nos complexes au bore avec des hydrosilanes et hydroboranes, nous avons démontré la coupure hétérolytique des liaisons Si–H et B–H, conduisant à des complexes silyl- et boryl-diazénido cationiques (Angew. Chem. Int. Ed. 2017 et Chem. Asian. J. 2024). Le diazote y est ainsi fonctionnalisé de manière contrôlée via des réactifs faiblement électrophiles.
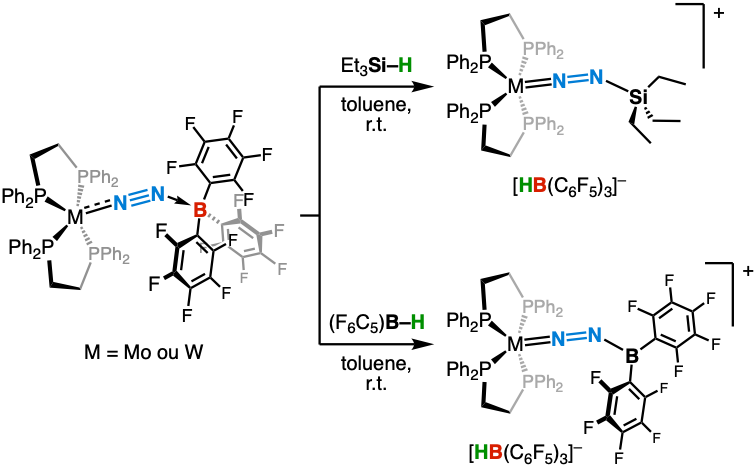
Formation de liaison entre le diazote activé et le silicium ou le bore.
Cette approche a été étendue à des complexes nitrure de molybdène, produits par scission du N₂, soulignant le potentiel des systèmes à franchir les étapes clefs d’un cycle de fixation. De plus, nous avons mis en évidence une borylation catalytique de N₂ par le borane HB(C₆F₅)₂, via un mécanisme PLF. Dans le cas de complexes de tungstène riches en électrons, cette activation s’accompagne d’une addition formelle 1,3 de la liaison B–H sur le fragment M–N≡N (Chem. Eur. J. 2019 et Dalton Trans. 2021).
Ces résultats confortent le rôle important des interactions PLF dans la valorisation du diazote, et ouvrent la voie à une fonctionnalisation catalytique de N₂ à l’échelle moléculaire.
Au-delà des stratégies classiques de scission du diazote – chimiosorption sur métal (Haber-Bosch) ou activation par deux centres métalliques riches en électrons – notre équipe a exploré une troisième voie : l’activation de N₂ via des systèmes donneurs-accepteurs extrêmes.
Nous avons utilisé un super-acide de Lewis bifonctionnel comportant deux centres bore fortement électrophiles. Une étude combinée (structures cristallines, spectroscopie, DFT) a montré que la double coordination sur N₂ engendre une polarisation si marquée qu’elle conduit à une réduction à deux électrons du ligand diazote. Cette activation profonde se manifeste par un abaissement de l’ordre de liaison de 3 à 2, marquant une étape clé vers sa rupture hétérolytique (Chem. Sci. 2023).
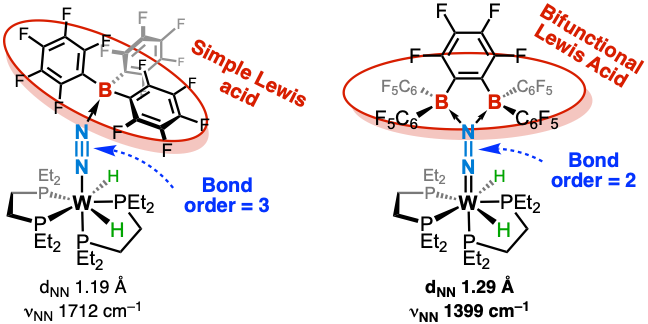
Vers la coupure hétérolytique de N2
Ces résultats ouvrent une nouvelle piste vers des mécanismes de réduction du diazote, illustrant tout le potentiel des architectures donneur-accepteur pour dépasser les paradigmes actuels de la fixation de N₂.
Faire réagir le diazote avec des molécules organiques riches en électrons : mythe ou réalité ?
Contrairement au monoxyde de carbone (CO), qui réagit classiquement avec des nucléophiles, le diazote (N₂) agit plutôt comme un nucléophile terminal lorsqu’il est lié à un métal de transition, réagissant avec des électrophiles grâce à une densité électronique accrue transmise par le métal. Cette réactivité opposée à celle du CO soulève un intérêt fondamental, notamment dans le contexte de la fixation de l’azote.
Une série de publications anciennes sur le complexe [CpMn(CO)₂N₂] rapportait l’addition d’organolithiens sur le ligand N₂. Toutefois, faute de reproduction indépendante, la véracité de cette transformation était sujette à caution.
Nous avons réévalué cette problématique à partir de complexes bis(N₂) du groupe 6. Aucune attaque directe du N₂ n’a été observée avec les organolithiens, mais le méthyllithium permet la substitution d’un ligand N₂ par un groupe méthyle, formant des complexes trans-méthyl-N₂ anioniques (Eur. J. Inorg. Chem. 2020).
Ces résultats nous ont amenés à revisiter expérimentalement la réactivité du complexe [CpMn(CO)₂N₂]. Grâce à des techniques modernes (RMN, diffraction RX, infrarouge in situ), nous avons démontré que l’organolithien PhLi réagit non pas sur N₂ mais sur un ligand CO, générant un complexe acyl-manganate instable [CpMn(CO)(COPh)N₂]⁻. Ce dernier se décompose à basse température en [CpMn(CO)₂Ph]⁻, erronément identifié dans l’étude historique comme un dérivé phényl-azo (Angew. Chem. Int. Ed. 2023).
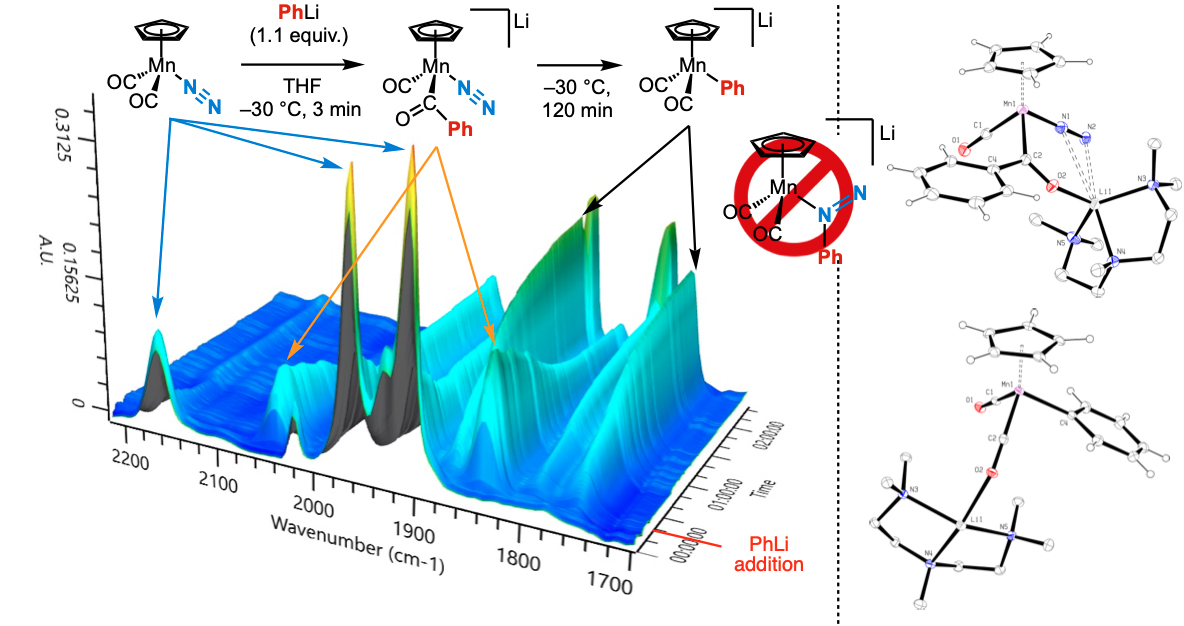
Suivi par infrarouge et caractérisation par diffraction des rayons X des produits formés par réaction entre un complexe de diazote et un nucléophile, le phényllithium.
Nos résultats, soutenus par des calculs DFT, clarifient un point longtemps ambigu de la chimie du diazote et renforcent l’importance d’une approche critique et structurée dans l’étude de ces systèmes hautement sensibles.
LCC CNRS
Laboratoire de chimie de coordination du CNRS
205 route de Narbonne, BP 44099
31077 Toulouse cedex 4
France
+ 33 5 61 33 31 00
