LCC
Pourquoi s’intéresser à la liaison carbone–fluor ?
La liaison C–F est l’une des plus fortes rencontrée en chimie organique, ce qui en fait à la fois un atout et un défi. Présente dans de nombreux composés fluorés industriels — médicaments, agrochimiques, matériaux — sa stabilité exceptionnelle rend ces molécules durables, parfois trop : leur accumulation dans l’environnement soulève des préoccupations sanitaires majeures. Activer ou rompre sélectivement la liaison C–F représente donc un enjeu scientifique important pour développer des voies de dégradation contrôlée, de recyclage, ou même de valorisation de ces composés.
Au-delà des applications environnementales, la fonctionnalisation de la liaison C–F ouvre la voie à la modification fine de molécules fluorées, permettant d’en ajuster les propriétés (réactivité, polarité, bioactivité). En s’attaquant à cette liaison réputée inerte, la chimie moderne repousse les limites de la catalyse, de la synthèse organométallique et de la chimie durable.
Nos recherches sur l’activation de la liaison carbone–fluor
Fonctionnalisation C–F par « umpolung » du cyclohexyne
Dans le cadre d’un projet européen ITN Marie Skłodowska-Curie, nous avons collaboré avec l’Université de York (Dr. John Slattery et Prof. Jason Lynam) pour développer une stratégie originale de fonctionnalisation de liaisons C–F via une inversion de polarité du cyclohexyne. Par coordination au zirconocène dans [Cp₂Zr(PMe₃)(c-C₂(CH₂)₄)], ce motif hautement contraint devient nucléophile et peut réagir sur des polyfluoropyridines (Chem. Sci. 2025).
Avec les 2,4,6-tri- et 2,6-difluoropyridines, une attaque du cyclohexyne sur l’hétéroaromatique forme des complexes 1-zircona-2-aza-cyclopentadiènes, aboutissant à une fonctionnalisation régiosélective de la liaison C–F. Les calculs DFT (Dr. John Slattery and Prof. Jason Lynam) confirment un mécanisme via un intermédiaire de type Meisenheimer, suivi d’une migration du fluor vers le zirconium.
Cette réactivité n’a pas été observée avec la 2,3,5,6-tétrafluoropyridine, probablement en raison d’une répulsion stérique et d’une stabilisation thermodynamique de la liaison M–C résultant d’un mécanisme d’activation de la liaison C–H en position 4. Enfin, la réaction avec la pentafluoropyridine a conduit à un mélange complexe, incluant des produits issus d’interactions parasites entre la PMe₃ et l’hétérocycle, nous orientant vers une nouvelle étude de la réactivité des trialkylphosphines avec les arènes fluorés.
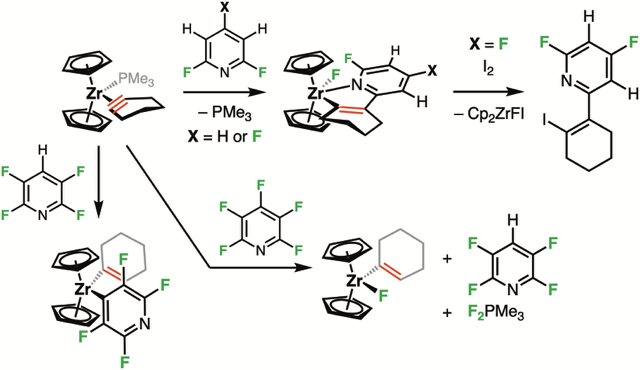
Diverses réactivités d’un complexe zirconium-cyclohexyne envers les pyridines fluorées
Activation C–F métallomimétique par les trialkylphosphines
Ces recherches nous ont par la suite permis de développer l’hydrodefluoration sur un panel restreint d’aromatiques fluorés (dès lors que ceux-ci sont fortement activés envers l’attaque nucléophile de la phosphine, première étape du mécanisme) en conditions catalytiques en phosphine. Notamment, la phosphine bon marché P(n-Bu)3 s’est révélée être notre meilleur catalyseur. Les calculs DFT sur cette transformations (Dr. John Slattery et Pr. Jason Lynam) et les investigations mécanistiques suggèrent que la phosphine cycle entre les états d’oxydation III et V, via des étapes d’additions oxydante, transmétallation et élimination réductrice, ce qui jusqu’alors n’avait été démontré, dans le cas des éléments du groupe 15, qu’avec des espèces possédant une sphère de coordination contrainte éloignant l’élément de sa géométrie favorite telle que dictée par la théorie VSEPR et permettant une diminution de l’écart HO–BV. Ces résultats suggèrent donc qu’une réactivité métallomimétique pour les éléments du groupe 15 peut être atteinte mettant en jeu des systèmes plus simples, et s’inscrivent plus largement dans le domaine de la chimie visant à remplacer les métaux par des éléments plus abondants en catalyse (J. Am. Chem. Soc. 2024).
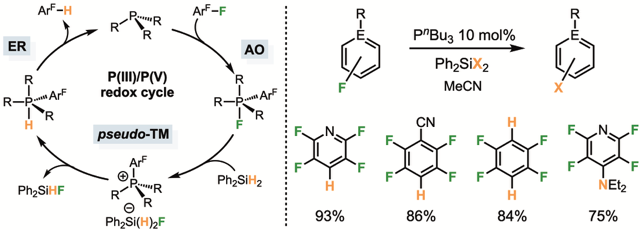
Développement d’une catalyse d’hydrodéfluoration métallomimétique catalysée par des phosphines commerciales.
LCC CNRS
Laboratoire de chimie de coordination du CNRS
205 route de Narbonne, BP 44099
31077 Toulouse cedex 4
France
+ 33 5 61 33 31 00
