LCC
Modélisation du phénomène de la transition de spin : Objectif et stratégies
L’objectif premier de la composante théorique de l’équipe est de simuler, de reproduire et d’interpréter les différents résultats expérimentaux observés dans l’équipe. Les principales méthodes théoriques employées sont des approches de physique statistique dites « hamiltoniennes » et la thermodynamique axiomatique incluant la thermomécanique (la mécanique des milieux continu des systèmes ouverts). L’une des préoccupations majeures est d’être capable de retrouver les bons ordres de grandeurs de quantités physiques extraites expérimentalement (propriétés mécaniques, thermodynamiques, vibrationnelles…) ou de déterminer les ingrédients physiques pertinents à l’origine du phénomène observé. La démarche théorique est donc à la fois soucieuse de porter un œil attentif aux observations expérimentales afin de donner une analyse quantitative et rationnelle des phénomènes étudiés et de déterminer les mécanismes physico-chimiques sous-jacents associés à ces phénomènes.
La première approche consiste à développer des modèles dits microscopiques ou atomistique (modèles de type Ising, modèles atome-phonon, systèmes masse-ressort complexes…) qui sont étudiés soit par des méthodes semi-analytiques ou résolus par des méthodes numériques (méthodes Monte Carlo ou/et dynamique moléculaire). Une seconde approche consiste à considérer des modèles mésoscopiques (mécanique des milieux continus, théorie de la réponse linéaire d’Onsager…) ou bien macroscopiques (thermodynamiques). Le réel avantage de ces modèles réside dans le fait que leurs paramètres sont en général des grandeurs physiques fondamentales mesurables (modules élastiques, vitesses du son, température de Debye, chaleur latente, capacité calorifique…).
Trois thématiques de recherche fortement interconnectées intéressent plus particulièrement la composante théorique de l’équipe :
- L’étude des mécanismes nucléation-croissance de domaine au cours du changement d’état de spin (dynamique spatio-temporelle). Analyse des critères de nucléation, des vitesses de propagation des parois de domaines… Simulation de la réponse optique et thermoélastique d’un matériau ou nanomatériau à transition de spin suite à l’application d’un laser pulsé (dynamique multi-échelle). Etude du temps de relaxation vers l’équilibre thermodynamique. Modélisation des expériences de microscopie pompe-sonde résolues en temps. Développement de nouveaux algorithmes Monte Carlo.
- Les effets de réduction de la taille sur les propriétés de commutation des matériaux à transition de spin (stabilité des phases, bistabilité). Modélisation des propriétés physico-chimiques de surface/interface et détermination quantitative des observables caractérisant les surfaces (énergies et contraintes de surface/interface). Simulation de la dynamique de réseau par des méthodes de dynamique moléculaire non conservative. Développement de champs de force intégrant le caractère commutable des composés à transition de spin (dynamique moléculaire du champ de ligand.)
- L’étude quantitative des propriétés mécaniques des matériaux à transition de spin sous diverses formes (cristaux, nanoparticules, films minces, systèmes cœur-coquille…) ainsi que des matériaux composites chargés par des nano-objets à transition de spin. Optimisation de structures et prédiction de synergies entre matrice et particules en vue de leur intégration dans des dispositifs de type actionneurs mécaniques (actuateurs) ou pour des applications en biomimétisme (muscles artificiels).
Highlight 1
“Role of Surface Effects in the Vibrational Density of States and the Vibrational Entropy in Spin Crossover Nanomaterials: A Molecular Dynamics Investigation”
Fahs, W. Nicolazzi, G. Molnár, and A. Bousseksou, Magnetochemistry, 7, (2021), 27
L’étude des propriétés vibrationnelles et mécaniques des matériaux à transition de spin peut être réalisée par des méthodes de dynamique moléculaire décrivant les matériaux à transition de spin à l’échelle atomique (figure 1 a). Les différentes interactions intra- et inter- moléculaires peuvent être modélisées par des champs de force dont les paramètres (constantes de forces, paramètres structuraux) peuvent être estimées à partir de données spectroscopiques (Raman, UV visible…) ou par diffraction des rayons X.
Cette méthode numérique permet d’extraire des grandeurs macroscopiques à partir de considérations microscopiques (description à l’échelle de la liaison). En particulier, ce travail a permis de simuler la dynamique de réseau dans les deux états de spin (figure 1 b) et de calculer de manière quantitative des grandeurs mécaniques ou thermodynamiques telles que le module d’Young (vitesse du son), l’entropie ou l’énergie interne vibrationnelle, quantités dont la connaissance est indispensable à la compréhension des mécanismes mis en jeu dans les phénomènes observés dans les matériaux à transition de spin.
Cette technique numérique a permis également permis d’appréhender les effets de la réduction de la taille sur la densité d’états vibrationnels de films minces à transition de spin. Elle a permis de montrer en autre que la diminution de l’épaisseur du film mince conduisait à l’émergence de modes de surface (modes dits de Rayleigh) dans la densité d’états vibrationnels. Ces modes additionnels affectent à la fois les propriétés mécaniques et entropiques des films minces corroborant les observations expérimentales obtenues par des mesures de diffusion inélastique nucléaire.
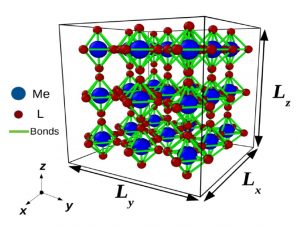
Figure 1 : a) Structure simplifiée d’un film mince à transition de spin : un réseau cubique à motifs octaédrique où chaque atome représentant soit le centre métallique (en bleu) ou les ligands (en rouge) est connecté par des liaisons (en vert) décrites par un champ de forces (énergies potentielles) approprié.
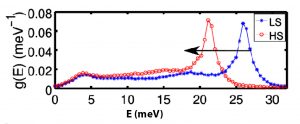
Figure 1.b) Calcul de la densité partielle d’états vibrationnels dans les deux états de spin par transformée de Fourier de la fonction d’autocorrélation des vitesses obtenue par simulations de dynamique moléculaire non-conservative. La flèche indique le ramollissement des modes optiques (mode d’élongation Métal-ligand) observé lors de la transition HS→LS.
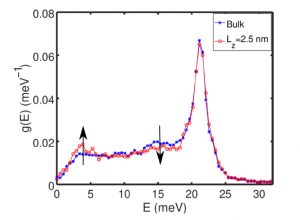
Figure 2 : Simulation de l’effet de la réduction de l’épaisseur d’une couche mince à transition de spin sur la densité d’états vibrationnels. Les flèches noires indiquent l’apparition de nouveaux modes de vibrations attribués à la présence de surfaces.
Highlight 2
“Influence of the ultra-slow nucleation and growth dynamics on the room-temperature hysteresis of spin-crossover single crystals” chem. Phys. Lett. 770, (2021), 138442.
Etude de la dynamique spatio-temporelle de la transition de spin thermique du composé [Fe(1,6-naphthy)2(Ag(CN)2)2] (1) (1,6-naphthy = 1,6-naphthyridine) par microscopie optique, permettant de suivre le processus de nucléation, la croissance de domaines et la propagation des parois de domaines au cours du changement d’état de spin. Ce composé présente une dynamique spatio-temporelle singulière en comparaison avec les observations couramment faites sur les monocristaux à transition de spin, attribuées à la présence d’une forte densité de défauts, microstructures et inhomogénéités. Les modèles de type Ising compressible, résolus par méthodes Monte Carlo, permettent de reproduire qualitativement les résultats de microscopiques optiques (Figure 3).
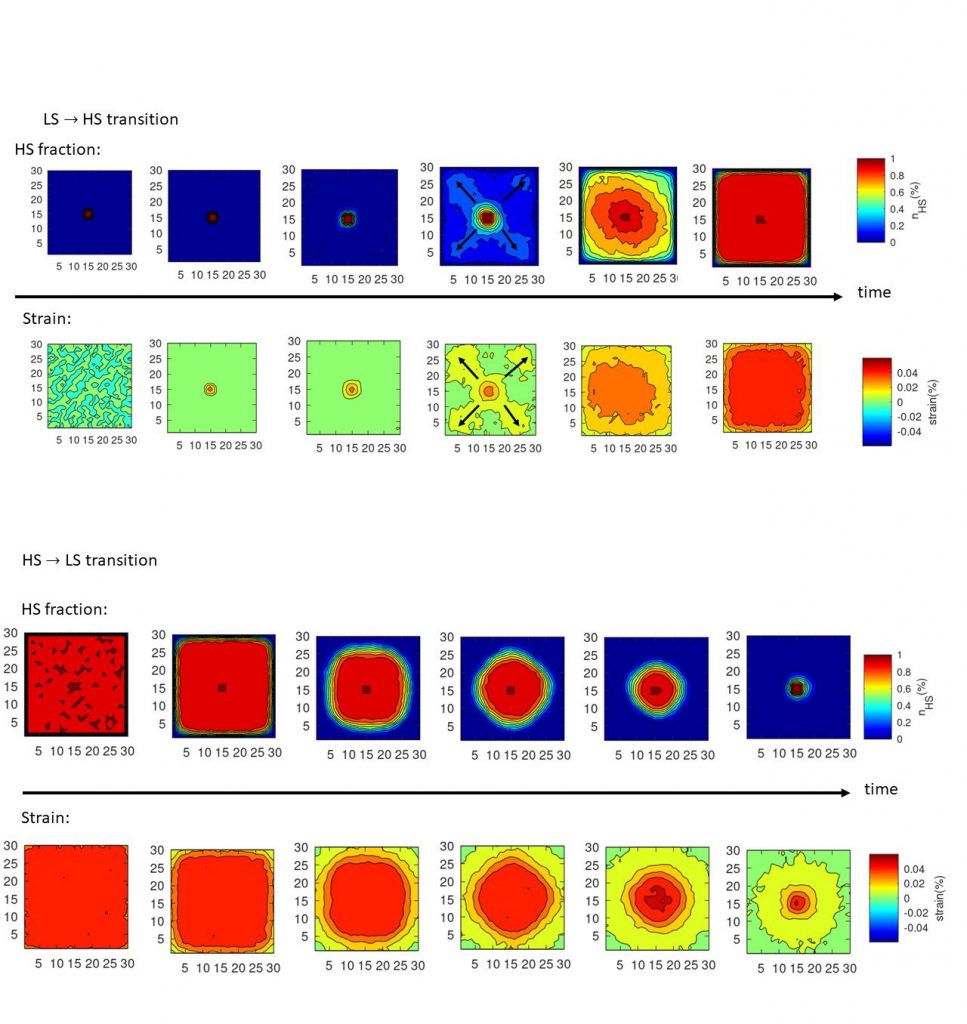
Figure 3 : Simulations de Monte Carlo montrant les distributions spatiales de la fraction HS locale moyenne et la trace du tenseur de déformation pendant les relaxations isothermes LS à HS (panneau supérieur) et HS à LS (panneau inférieur)
LCC CNRS
Laboratoire de chimie de coordination du CNRS
205 route de Narbonne, BP 44099
31077 Toulouse cedex 4
France
+ 33 5 61 33 31 00
