LCC
Atmospheric nitrogen: an inert yet essential resource
Molecular nitrogen (N₂) makes up nearly 80% of the air we breathe. Yet this molecule is remarkably inert due to its strong triple bond, making it extremely difficult to activate. In nature, only certain microorganisms are capable of “fixing” nitrogen, converting it into ammonia (NH₃)—a reactive form essential for building life’s key components, such as amino acids. This transformation is catalyzed by enzymes known as nitrogenases. Since the 20th century, industry has developed a similar process—the Haber-Bosch process—to produce ammonia on a large scale for use in fertilizers. However, this process is highly energy-intensive and emits large amounts of CO₂. Inspired by nitrogenases, chemists are now working to design new materials and molecular catalysts capable of fixing nitrogen under milder and more sustainable conditions.
Our research on nitrogen activation
Dinitrogen activation & functionalization through donor–acceptor systems
Our team develops heterobimetallic strategies for N₂ activation, drawing inspiration from nitrogenase enzymes. The approach involves coupling an electron-rich metal center (group 6 M⁰ complexes) with a strong Lewis acid, allowing for polarization of the N₂ ligand and the emergence of unprecedented reactivity.
We showed that coordination of B(C₆F₅)₃ to the N₂ ligand induces elongation of the N≡N bond and a significant decrease in its vibrational frequency. The N≡N–B angle (~140°) reflects effective overlap between the vacant p orbital of boron and the π* orbital of the diazene ligand (Angew. Chem. Int. Ed. 2017).
We also investigated cationic Au(I) fragments, both for their ability to promote N–C bond formation and as isolobal analogues of H⁺. Spectroscopic and DFT studies indicate that N₂ activation in these systems relies on strong electrostatic effects, suggesting a key role in the enzymatic reduction of N₂ (Inorg. Chem. 2021).
With Al(C₆F₅)₃, although steric hindrance slightly reduces activation efficiency, we isolated doubly activated adducts (M{–N≡N–Al(C₆F₅)₃}₂), elusive with boron-based systems. DFT calculations further reveal a decrease in the HOMO–LUMO gap, indicating improved electron/proton transfer properties—essential traits for catalysis (Chem. Sci. 2024).
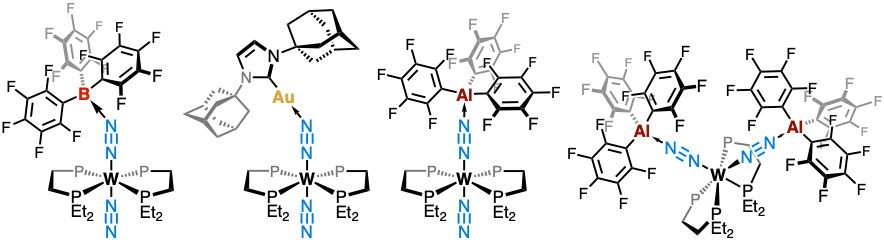
Dinitrogen activation with tungsten combined to boron, gold or aluminium.
Our work thus paves the way for homogeneous N₂ fixation assisted by Lewis acids, acting as co-catalysts in mild redox cycles closer to biological conditions.
Our M–N₂/Lewis acid adducts behave like frustrated Lewis pairs (FLPs), capable of activating strong bonds under mild conditions. By reacting our boron-based complexes with hydrosilanes and hydroboranes, we demonstrated heterolytic cleavage of Si–H and B–H bonds, leading to silyl- and boryl-diazene cationic complexes (Angew. Chem. Int. Ed. 2017 and Chem. Asian. J. 2024). This results in controlled functionalization of N₂ using weak electrophiles.
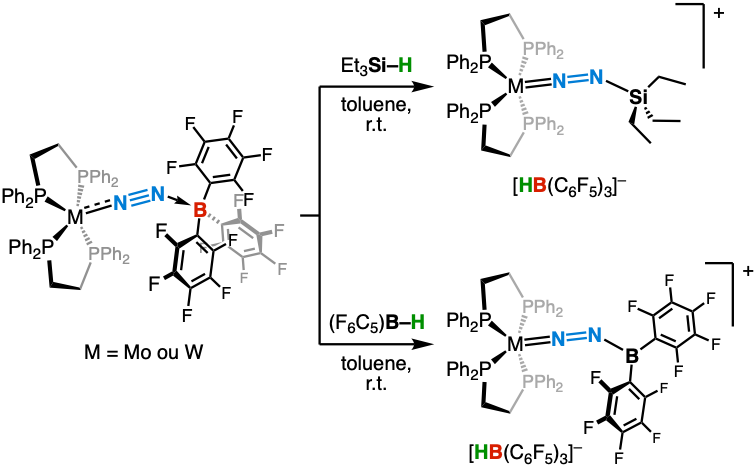
Chemical bond formation between activated dinitrogen and silicon or boron
We extended this approach to molybdenum nitride complexes—generated by N₂ splitting—highlighting the potential of such systems to progress through key steps of the fixation cycle. Moreover, we demonstrated catalytic borylation of N₂ with HB(C₆F₅)₂ via an FLP-type mechanism. In the case of electron-rich tungsten complexes, this activation proceeds via formal 1,3-addition of the B–H bond across the M–N≡N fragment (Chem. Eur. J. 2019 and Dalton Trans. 2021).
These findings underscore the central role of FLP-type interactions in N₂ valorization and open the door to catalytic molecular-scale N₂ functionalization.
Beyond conventional N₂ cleavage strategies—chemisorption on metal surfaces (Haber–Bosch) or activation by two electron-rich metal centers—our team explored a third route: extreme donor–acceptor activation of N₂.
We employed a bifunctional Lewis superacid featuring two highly electrophilic boron centers. Combined structural (XRD), spectroscopic, and DFT studies revealed that dual coordination to N₂ induces such strong polarization that the ligand undergoes a formal two-electron reduction. The resulting decrease in bond order (from 3 to 2) marks a critical step toward heterolytic N≡N bond cleavage (Chem. Sci. 2023).
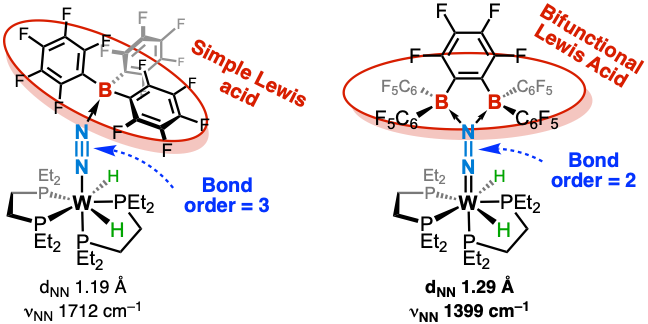
Toward the heterolytic cleavage of N2
These results suggest a new direction for N₂ reduction mechanisms, highlighting the full potential of donor–acceptor architectures in overcoming current limitations in N₂ activation.
N₂ reaction with nucleophilic organic species—myth or reality?
Unlike CO, which typically reacts with nucleophiles, metal-bound N₂ behaves as a nucleophile, reacting instead with electrophiles due to enhanced terminal electron density. This inversion of reactivity raises fundamental interest in the context of N₂ functionalization.
A series of early reports on [CpMn(CO)₂N₂] described nucleophilic addition of organolithium reagents to the N₂ ligand. However, the lack of independent reproduction cast doubt on the validity of these transformations.
We revisited this question using group 6 bis(N₂) complexes. While no direct attack on N₂ was observed, substitution of an N₂ ligand by a methyl group occurred with MeLi, yielding trans-methyl–N₂ anionic complexes (Eur. J. Inorg. Chem. 2020).
These findings led us to reinvestigate the original [CpMn(CO)₂N₂] chemistry. Using modern techniques (NMR, XRD, in situ IR), we demonstrated that PhLi actually reacts with a CO ligand, forming a thermally sensitive acyl-manganate [CpMn(CO)(COPh)N₂]⁻, which decomposes below –20 °C into [CpMn(CO)₂Ph]⁻. The latter had been misassigned in the original work as a phenyl-azo complex (Angew. Chem. Int. Ed. 2023).
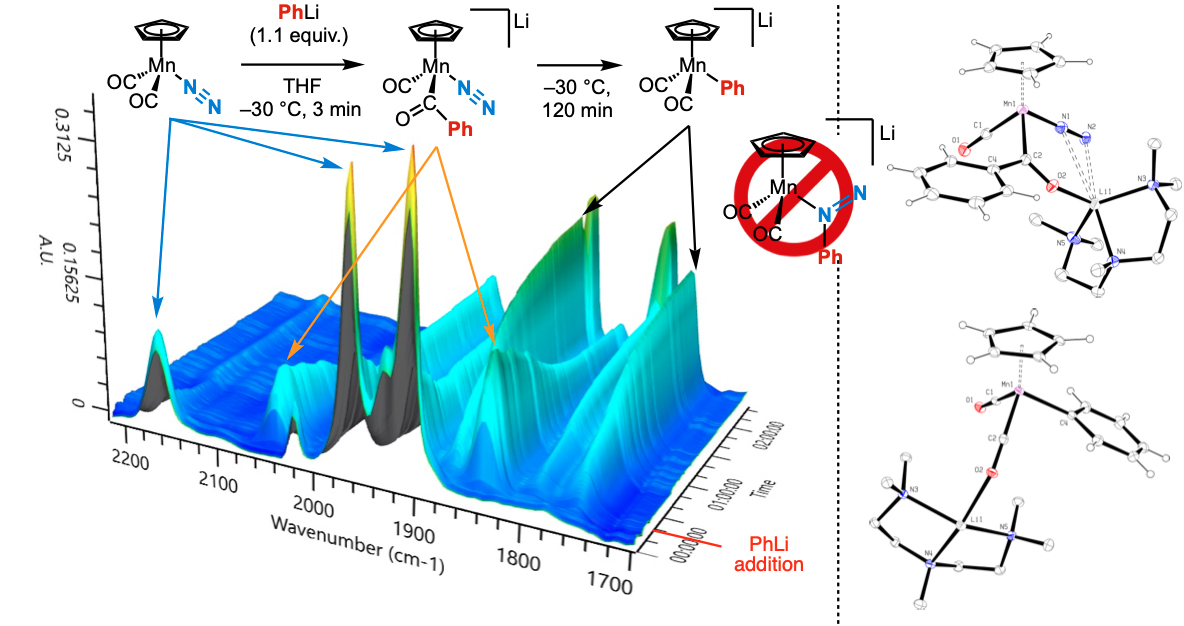
Infrared monitoring and X-ray diffraction characterization of products formed by reaction between a dinitrogen complex and a nucleophile, phenyllithium.
These results, supported by DFT, clarify a long-standing ambiguity in N₂ chemistry and emphasize the importance of critical re-evaluation and robust characterization in the study of reactive dinitrogen complexes.
LCC CNRS
Laboratoire de chimie de coordination du CNRS
205 route de Narbonne, BP 44099
31077 Toulouse cedex 4
France
+ 33 5 61 33 31 00
