LCC
Why Study the Carbon-Fluorine Bond?
The C–F bond is one of the strongest in organic chemistry, making it both an asset and a challenge. Found in many industrial fluorinated compounds—including pharmaceuticals, agrochemicals, and materials—its exceptional stability leads to persistent molecules, sometimes too persistent: their accumulation in the environment raises serious health concerns. Selective activation or cleavage of the C–F bond is therefore a key scientific challenge for developing pathways for controlled degradation, recycling, or valorization.
Beyond environmental applications, C–F bond functionalization enables fine-tuning of fluorinated molecules, allowing modulation of properties such as reactivity, polarity, and bioactivity. By targeting this notoriously inert bond, modern chemistry pushes the boundaries of catalysis, organometallic synthesis, and sustainable chemistry.
Our Research on Carbon-Fluorine Bond Activation
C–F Functionalization via Cyclohexyne Polarity Inversion
As part of a Marie Skłodowska-Curie ITN project, we collaborated with the University of York (Dr. John Slattery and Prof. Jason Lynam) to develop a novel strategy for C–F bond functionalization via polarity inversion of cyclohexyne. Upon coordination to zirconocene in [Cp₂Zr(PMe₃)(c-C₂(CH₂)₄)], this highly strained motif becomes nucleophilic and reacts with polyfluoropyridines (Chem. Sci. 2025). With 2,4,6-tri- and 2,6-difluoropyridines, nucleophilic attack of cyclohexyne on the heteroaromatic ring forms 1-zircona-2-aza-cyclopentadiene complexes, resulting in regioselective C–F bond functionalization. DFT calculations (by Dr. Slattery and Prof. Lynam) support a mechanism involving a Meisenheimer-type intermediate followed by fluorine migration to zirconium. This reactivity was not observed with 2,3,5,6-tetrafluoropyridine, likely due to steric repulsion and thermodynamic stabilization of the M–C bond resulting from C–H activation in 4-position. Reaction with pentafluoropyridine led to a complex mixture, including 1-cyclohexenylfluorozirconocene and side products from undesired interactions between PMe₃ and the heterocycle, prompting further study into the reactivity of trialkylphosphines with fluorinated arenes.
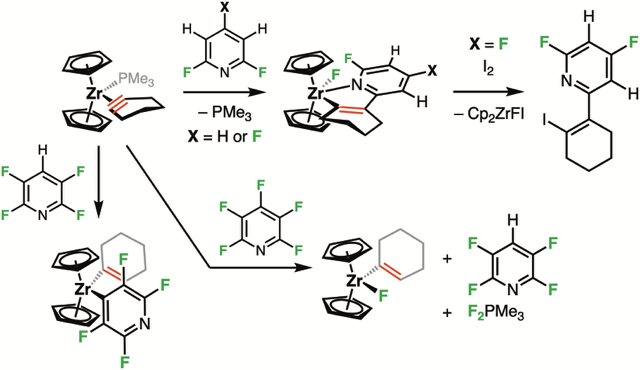
Various Reactivities of a Zirconium–Cyclohexyne Complex with Fluoropyridines.
Metallomimetic C–F Activation by Trialkylphosphines
These studies subsequently enabled the development of hydrodefluorination reactions on a limited set of fluorinated aromatics, provided they are strongly activated toward nucleophilic attack by phosphines—the first step in the mechanism. Notably, the inexpensive phosphine P(n-Bu)₃ proved to be the most effective catalyst. DFT calculations and mechanistic investigations (by Dr. Slattery and Prof. Lynam) suggest that the phosphine cycles between oxidation states III and V, via oxidative addition, transmetallation, and reductive elimination steps. Until now, such a mechanism had only been demonstrated for group 15 elements bearing highly constrained coordination spheres that deviate from their preferred VSEPR geometry and lower the HOMO–LUMO gap. Our results thus show that group 15 elements can exhibit metal-like reactivity using simpler systems, contributing to the broader goal of replacing transition metals with more abundant elements in catalysis (J. Am. Chem. Soc. 2024).
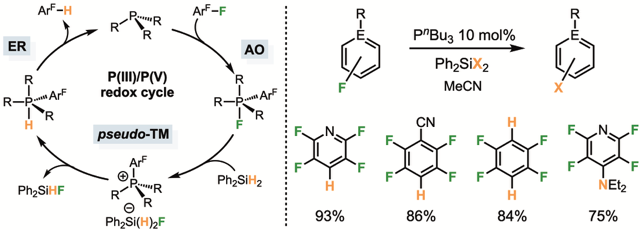
Metallomimetic hydrodefluorination catalyzed by simple phosphines.
LCC CNRS
Laboratoire de chimie de coordination du CNRS
205 route de Narbonne, BP 44099
31077 Toulouse cedex 4
France
+ 33 5 61 33 31 00
